





Codes
Some codes that I developed:
-
Surface Atoms Analysis
This python script analyzes which atoms of a molecule (in xyz file, for instance) are exposed to the vacuum and is exposed area.
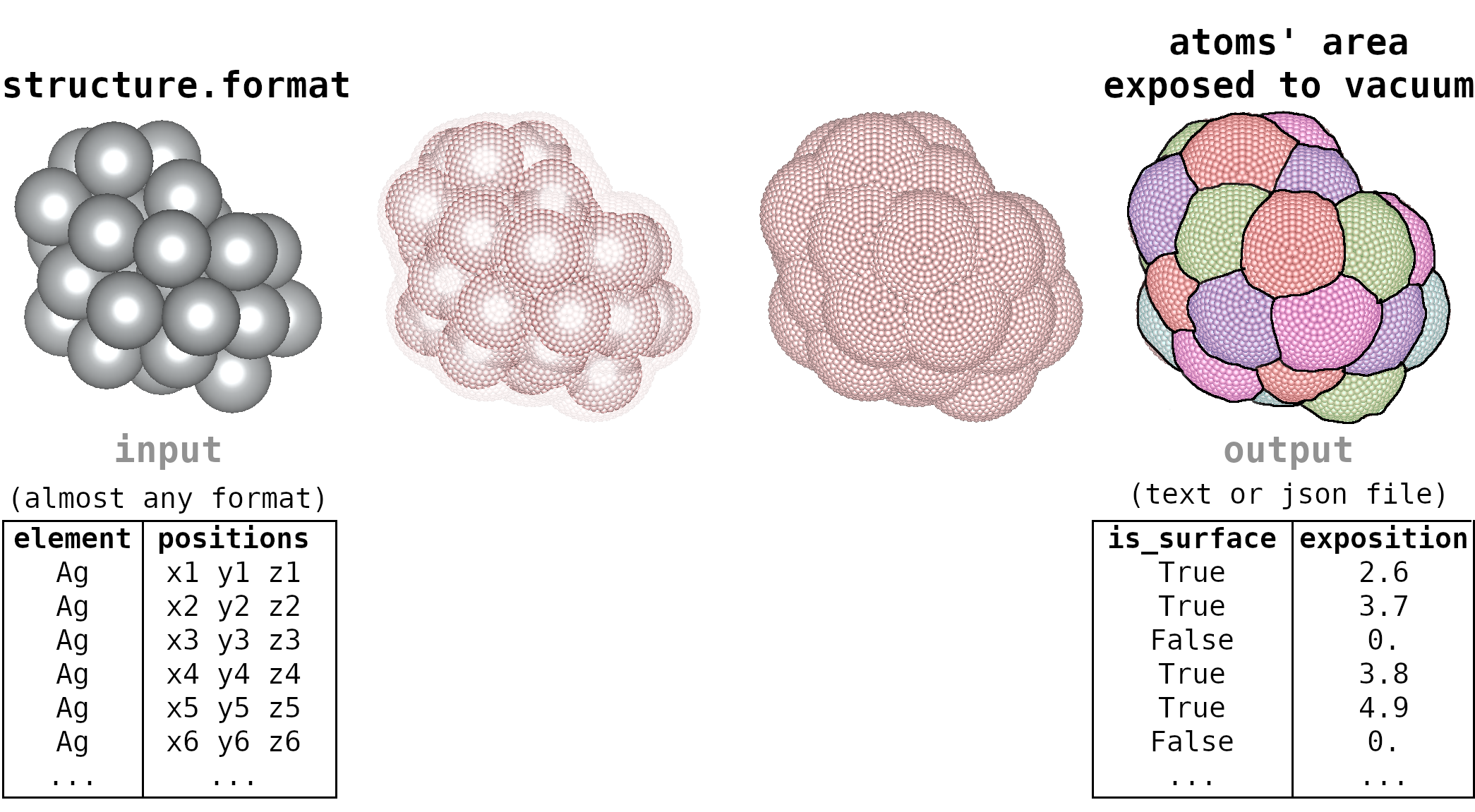
The atoms exposed to the vacuum could be considered surface atoms and often play important roles in the physical-chemical properties of small clusters/nanoclusters/nanostructures. I strongly recommend this analysis for the investigations of structures with more than 20 atoms.
I developed and implemented this methodology. You can find a detailed description in Section V of this document.
-
Effective Coordination Number
For each atom i, the effective coordination number (ECNi) and the average bond distance (davi) are defined as:
![ECN^{i} = \sum_{j \neq i}^{M} P_{ij},\;\;\;\;\;\;d^{i}_{av} = \dfrac{ \sum\limits_{j \neq i}^{M} d_{ij}P_{ij} }{ \sum\limits_{j \neq i}^{M} P_{ij} },\;\;\;\;\;\;P_{ij} = \exp \left[ 1- \left(\dfrac{2d_{ij}}{d_{av}^{i}+d_{av}^{j}}\right)^{6} \right]](/assets/images/Tex2Img_ecndavpij.png)
Where Pij is a function that weights the effective coordination between the atoms i and j.
Note that ECNi is a simple sum of Pij for between i and each other atom j. While davi is the sum of the distance between i and each other atom j, weighted by the Pij terms.
This python script calculates both the effective coordination number and average bond distance from almost any atomic structure file type. It works with any atomic structures file readable by ase (atomic simulation environment), including structures with periodic boundary conditions.
I just implemented this method which was developed by Da Silva, et al..
-
Quandarium
A python package that presents several tools to deal with Quantum Chemistry (QC) data of molecular systems (non-periodic):
- Extraction of data from QC calculations;
- Structural analysis, such as effective coordination bonds and surface atoms analysis (see below);
- AtoMF (Atomic to Molecular Featurization) (see more);
- Correlation and correlation significance analysis with bootstrap approaches.

The version presented here is an old version of Quandarium. I am developing the 1.0.0 version, which I expect to release soon, together with more tools and information about how to install and use it.