






1. About Me
I hold a bachelor's degree in Chemistry from the Federal University of Santa Catarina, which I obtained between 2013 and 2016. Subsequently, I pursued a Master's degree in Physical Chemistry at the São Carlos Institute of Chemistry, University of São Paulo, from 2016 to 2018. Throughout my undergraduate and master's studies, I focused on investigating various properties of organic molecules and oxide surfaces using the Density Functional Theory (DFT).
Since 2018, I have been pursuing a PhD at the São Carlos Institute of Chemistry, University of São Paulo. My current research is centered around data mining quantum chemistry datasets to extract valuable insights and knowledge. In addition to my doctoral studies, I am in the final stages of completing an MBA in Data Science and Analytics at Luiz de Queiroz College of Agriculture, University of São Paulo. As part of my MBA program, I conducted research utilizing Web Scraping to collect thousands of news and Natural Language Processing (NLP) techniques to analyze media bias.
During the first phase of my PhD, from 2018 to 2021, I worked under the supervision of Prof. Dr. Juarez L. F. Da Silva. I am now in the process of developing the second part of my PhD research under the guidance of Albérico Borges Ferreira da Silva.
Additionally, I engage in freelance work as a scientific illustrator, where I utilize my skills to visually communicate scientific concepts and ideas.
2. Education
PhD in Physical Chemistry (2018-Present)
- University of São Paulo (USP), São Carlos Institute of Chemistry, São Carlos, Brazil.
- Advisor (2021-Present): Albérico Borges Ferreira da Silva
- Advisor (2018-2021): Juarez L. F. Da Silva.
- Keywords: Atomistic Simulation, Data Mining, and Machine Learning.
MBA in Data Science and Analytics (2021-Present)
- University of São Paulo (USP), Luiz de Queiroz College of Agriculture, Piracicaba, Brazil.
- Advisor: Jéssica Peixoto de Araújo
- Term Paper (in Portuguese): Version sent to thesis defense committee.
- Dataset collected:

- Keywords: NLP, Text Mining, and Media Bias.
MSc in Physical Chemistry (2016-2018)
- University of São Paulo (USP), São Carlos Institute of Chemistry, São Carlos, Brazil.
- Advisor: Juarez L. F. Da Silva.
- Master thesis (in Portuguese): link to the repository.
- Keywords: Atomistic Simulation, Density functional Theory, CeO2, and CeO2-ZrO2.
BSc in Chemistry (2013-2016)
- Ferderal University of Santa Catarina, Department of Chemistry, Florianópolis, Brazil.
- Advisor: Giovanni F. Caramori.
- Undergraduate thesis (in Portuguese): link to the repository.
- Keywords: Cation-Π interaction, Π-Π interaction, and Transition metal complexes.
3. Work
Scientific illustration:
As a freelance scientific illustrator, I go by the artistic name Atomic Render.
You can check out my illustrations on Instagram @atomic.render 
For a detailed portfolio, please access my online portfolio ![]() ,
or download it
,
or download it  .
.
-
Art for presentations:

-
Animation based on paper result: Transition Metal clusters of Fe, Co, Ni, and Cu growing until 15 atoms. I processed and rendered the cluster's structures, which were previously developed in my research group (QTNano) by Dr. Anderson S. Chaves and coworkers (Phys. Chem. Chem. Phys., 2017,19, 15484-15502).
-
Graphical entry of the publication: Ab initio investigation of the role of the d-states occupation on the adsorption properties of H2, CO, CH4 and CH3OH on the Fe13, Co13, Ni13 and Cu13 clusters

Codes
-
Molecular Adsorption Algorithm by Surface Mapping (MAASM)

This algorithm generates sets of atomic structures of two adsorbed molecules, considering ridge structures and atoms as spheres of VDW radius.
My methods mimic the ideia that two melecules could interact based on different chemical environments on the surface of the molecules. First, it get a representative sets of chemical environments of each one of the molecules. Second, we find structures combining the molecules through each possible pair of chemical environments in the molecules. Finally, this poll of structures is sampled to find a representative set of thepossible ways of interaction.
-
Surface Atoms Analysis
This python script analyzes which atoms of a molecule (in xyz file, for instance) are exposed to the vacuum and is exposed area.
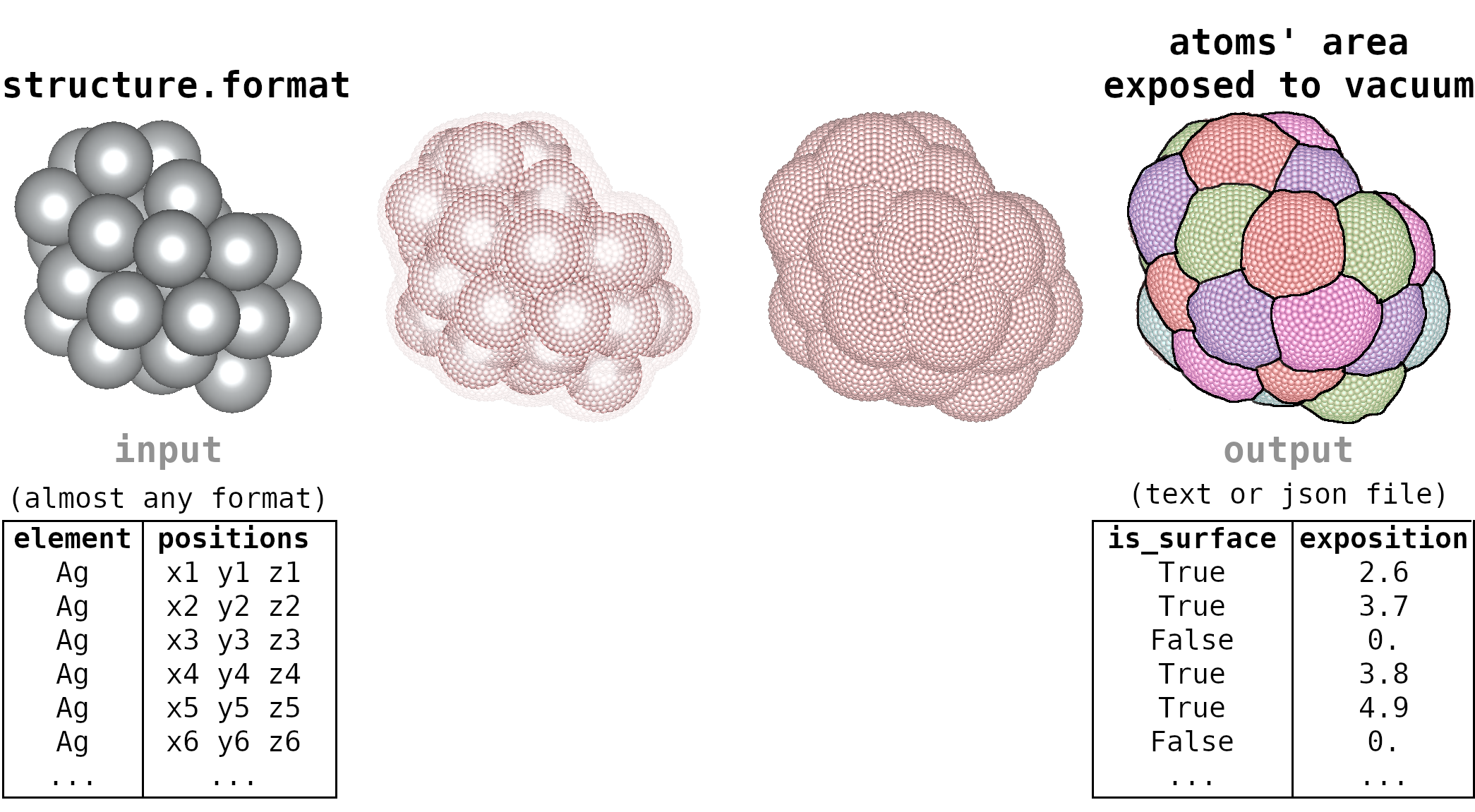
The atoms exposed to the vacuum could be considered surface atoms and often play important roles in the physical-chemical properties of small clusters/nanoclusters/nanostructures. I strongly recommend this analysis for the investigations of structures with more than 20 atoms.
I developed and implemented this methodology. Check a detailed description in Section V of this document.
-
Effective Coordination Number
This python script calculates, for each atom i, the effective coordination number (ECNi) and the average bond distance (davi), based on the weights the effective coordination (Pij) with other atoms j.
My implementation works with any atomic structures file readable by ase (atomic simulation environment), including structures with periodic boundary conditions. Method developed by Da Silva, et al..
-
Quandarium
A python package that presents several tools to deal with Quantum Chemistry (QC) data of molecular systems (non-periodic): extraction of data from QC calculations; structural analysis, such as effective coordination bonds and surface atoms analysis (see above); Atomic to Molecular Featurization (see above); correlation and correlation significance analysis with bootstrap approaches.
The version presented here is an old version of Quandarium. I am developing the 1.0.0 version, which I expect to release soon, together with more tools and information about how to install and use it.
Scientific Research:
Papers:
- Correlation-Based Framework for Extraction of Insights from Quantum Chemistry Databases: Applications for Nanoclusters. Journal of Chemical Information and Modeling, 2021, 61 (3), 1125-1135. DOI: 10.1021/acs.jcim.0c01267
- Machine Learning Prediction of Nine Molecular Properties Based on the SMILES Representation of the QM9 Quantum-Chemistry Dataset. The Journal of Physical Chemistry A, 2020, 124 (47), 9854-9866. DOI: 10.1021/acs.jpca.0c05969
- Methane dehydrogenation on 3d 13-atom transition-metal clusters: A density functional theory investigation combined with Spearman rank correlation analysis. Fuel, 2020, 275, 117790. DOI: 10.1016/j.fuel.2020.117790
- Ab Initio Insights Into the Formation Mechanisms of 55-Atom Pt-Based Core-Shell Nanoalloys. The Journal of Physical Chemistry, 2020, 124, 1, 1158-1164. DOI: 10.1021/acs.jpcc.9b09561
- Ab initio insights into the structural, energetic, electronic, and stability properties of mixed CenZr15-nO30 nanoclusters, Physical Chemistry Chemical Physics, 2019, 21, 26637-26646. DOI: 10.1039/C9CP04762J
- Understanding the interplay between π-π and cation-π interactions in [janusene-Ag]+ host-guest systems: a computational approach, Dalton Transactions, 2019, 48, 13281-13292. DOI: 10.1039/C9DT02307K
- Ab initio investigation of the formation of ZrO2-like structures upon the adsorption of Zrn on the CeO2(111) surface, The Journal of Chemical Physics, 2018, 149, 244702. DOI: 10.1063/1.5063732
- From Bulk CeO2 to Transition-Metal Clusters Supported on the CeO2(111) Surface: A Critical Discussion. In book: Encyclopedia of Interfacial Chemistry, Surface Science and Electrochemistry, 2018, 452-459. DOI: 10.1016/B978-0-12-409547-2.14196-4
Ongoing Researches:

The following papers employed AtoMF features:
I am working on developing models that facilitate the extraction of knowledge and the relationship between AtoMF features and the targets.
I am also working on anlysis
Talks & Hands-on:
- Open talk entitled "How Machine Learning and Chemistry Meet in the Quantum Chemistry Field", which occurred online in August 31, 2020.
- Open talk entitled "A Feature Engineering and Correlation-based Framework to Knowledge Extraction from Quantum Chemistry Datasets: the Nanoclusters Examples" in the V CINE-CMSC Workshop, which occurred online from July 06 to 10, 2020.
- Hands-on (3 days) entitled "Machine Learning Aplications and Introdution to Data Analysis with Python", in the IV CINE-CMSC Workshop: Machine Learning Techniques Applied to Computational Material Science with Hands-on, which occurred in São Carlos, São Paulo, from February 10 to 14, 2020.
- Open talk entitled "A Correlation and Feature Engineering Based Framework to Obtain Insights from Quantum Chemistry Datasets" in the IV CINE-CMSC Workshop: Machine Learning Techniques Applied to Computational Material Science with Hands-on, which occurred in São Carlos, São Paulo, from February 10 to 14, 2020.

- Open talk entitled "Data Mining and Statistical Tools based Framework to Investigate Quantum Chemistry Data" in the III CINE-CMSC Workshop, which occurred in São Carlos, São Paulo, from September 02 to 06, 2019.
- Hands-on (3 days) entitled "Tutorial on Vienna Ab initio Simulation Package (VASP)", in the II CINE-CMSC Workshop, which occurred in São Carlos, São Paulo, from February 13 to 15, 2019.
- Open talk entitled "Ab initio on investigation of Zr Adatoms on the CeO2(111) surface." in the III School of Computational Chemistry, which occurred in Ribeirão Preto, São Paulo, from December 11 to 14, 2017.
4. Resume:
Resume in portuguese:
Scientific illustration portfolio:
5. Contact
E-mail is the best way to get in touch with me.
- johnatanmucelini@usp.br
- johnatanmucelini@gmail.com